
 NAT法|支原体qPCR检测和方法验证
NAT法|支原体qPCR检测和方法验证
11100
2023-11-14
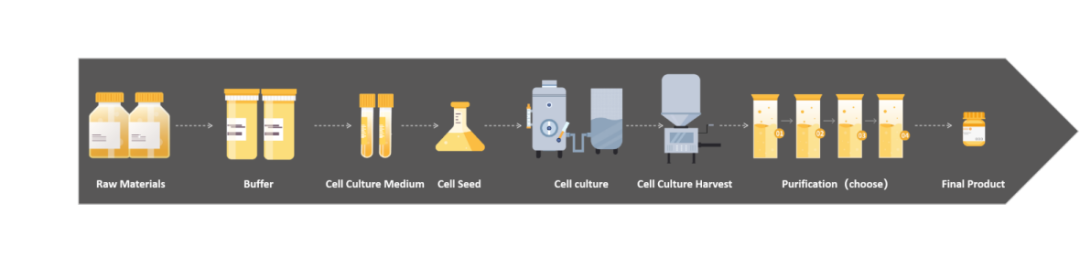
近些年,随着生物医药迅猛发展、疫情下细胞基因治疗以及mRNA疫苗的相继涌现,如何把控生物制品安全可靠,成为各国法规政府重视以及监管的要点。支原体是一种比较常见但通常难以去除的污染类型,对于涉及细胞培养的生物制品工艺过程,法规要求“必须确保无支原体污染”。
监管机构要求的支原体检测点
-
2020版中国药典三部《生物制品生产检定用动物细胞基质制备及质量控制》中提出对于生产用细胞,需要对主细胞库(MCB)、工作细胞库(WCB)、生产终末细胞(EOPC)进行支原体检查。
-
《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》中建议在关键时间点对适合的中间样品开展支原体等安全性相关检测或采取相关的措施加以控制,并且支原体同样需要作为终产品的放行检项。
-
FDA,Guidance for Industry:Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications,中提出对于原材料、病毒种子、未加工的收获液等都需要进行支原体控制。
NAT法和传统方法
NAT法-方法验证要求
专属性(Specificity)
检测限(Detection Limit)
|
机构 |
欧洲药典<2.6.7> |
日本药典<G3-14-170> |
美国药典<63> |
中国药典2020 <3301> |
WHO |
|
|
验证要求 |
专属性、检测限、耐用性 |
专属性、检测限、耐用性 |
与法定方法进行可比性研究 |
未明确(需要经国家药品检定机构认可) |
与法定方法进行可比性研究(包括专属性和检测限) |
|
|
需验证的支原体菌株 |
1 |
莱氏无胆甾原体 |
莱氏无胆甾原体 |
莱氏无胆甾原体 |
\ |
莱氏无胆甾原体 |
|
2 |
发酵支原体 |
发酵支原体 |
发酵支原体 |
\ |
发酵支原体 |
|
|
3 |
猪鼻支原体 |
猪鼻支原体 |
猪鼻支原体 |
猪鼻支原体*** |
\ |
|
|
4 |
口腔支原体 |
口腔支原体 |
口腔支原体 |
口腔支原体 |
口腔支原体 |
|
|
5 |
肺炎支原体 |
肺炎支原体 |
肺炎支原体 |
肺炎支原体 |
肺炎支原体 |
|
|
6 |
鸡毒支原体 |
\ |
鸡毒支原体 |
\ |
\ |
|
|
7 |
滑液囊支原体* |
滑液囊支原体* |
滑液囊支原体* |
\ |
\ |
|
|
8 |
精氨酸支原体 |
精氨酸支原体 |
\ |
\ |
\ |
|
|
9 |
螺原体** |
螺原体** |
\ |
\ |
\ |
|
|
10 |
\ |
唾液支原体 |
\ |
\ |
\ |
|
-
如果在生产过程中使用或接触禽细胞或材料,应进行滑液囊支原体的检测限验证;
-
如果在生产过程中使用或接触昆虫或植物材料,应进行螺原体的检测限验证;
-
猪鼻支原体在支原体污染样本中占比非常高,受到中国食品药品检定研究院的关注。
耐用性(Robustness)
可比研究(Comparability Study)
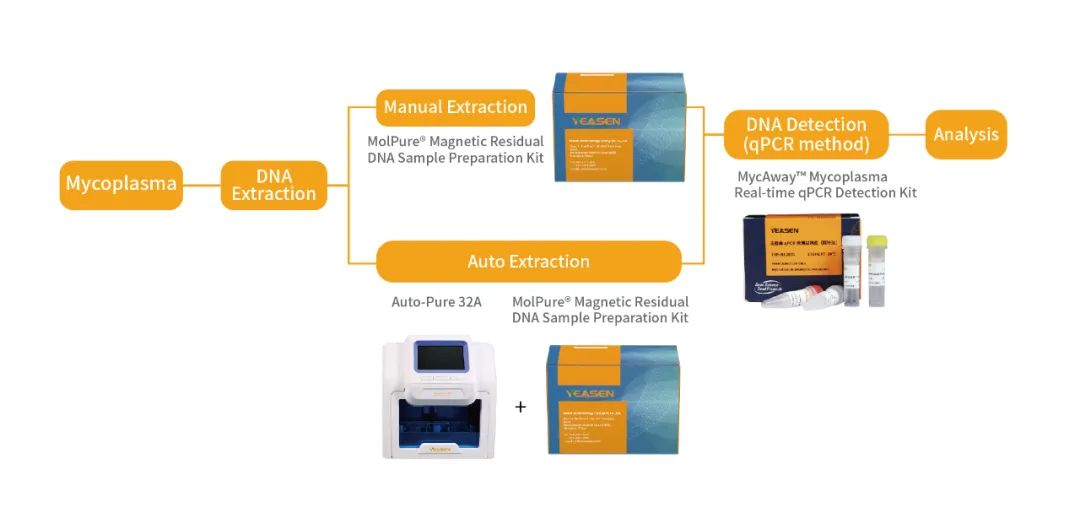
NAT法支原体qPCR检测——完整解决方案
相关产品
|
产品名称 |
货号 |
规格 |
|
MolPure® Magnetic Residual DNA Sample Preparation Kit 磁珠法残留DNA样本前处理试剂盒(瓶装) |
18461ES25 |
25 T |
|
MolPure® Magnetic Residual DNA Sample Preparation Kit 磁珠法残留DNA样本前处理试剂盒(瓶装) |
18461ES60 |
100T |
|
MolPure® Mag32 Residual DNA Sample Preparation Kit FA 磁珠法残留DNA样本前处理试剂盒FA(预封装) |
18462ES32 |
2×16 T |
|
MolPure® Mag32 Residual DNA Sample Preparation Kit FA 磁珠法残留DNA样本前处理试剂盒FA(预封装) |
18462ES59 |
6×16T |
|
Auto-Pure 32A automated nucleic acid extraction system Auto-Pure 32A 全自动核酸提取仪 |
80501ES01 |
32通量 |
|
MycAway™ Mycoplasma Real-time qPCR Detection Kit 支原体qPCR检测试剂盒(探针法) |
40618ES25 |
25 T |
|
MycAway™ Mycoplasma Real-time qPCR Detection Kit 支原体qPCR检测试剂盒(探针法) |
40618ES60 |
100T |
MycAway™ 支原体检测的验证内容
| 性能 | 验证参数 | 试剂盒验证结果 |
| 检测限 | 支原体菌株检测限 | 10种法规药典要求的支原体10CFU/mL,分别检测24次,检出率≥95%。 |
| 专属性 | 样本基质干扰 | 共测试9种样本基质无干扰(其中包括哺乳动物细胞培养基等) |
| 交叉反应 | 与21种常见的工程菌、工程细胞以及哺乳动物细胞基因组DNA无交叉反应 | |
| 耐用性 | 冻融稳定性 | 可经受反复5次冻融,试剂盒性能不受影响 |
| 热加速稳定性 | 37℃条件下10天和4℃条件下30天,试剂盒性能不受影响 | |
| 仪器适用性 | 适用ABI 7500、ABIQuantStudioTM 5、Bio-Rad CFX96 | |
| 覆盖度 | 覆盖支原体DNA种类 | 通过支原体16SrRNA序列进行数据库比对,覆盖包括支原体、螺原体和 无胆甾原体在内的160多种DNA序列。 |
技术服务
福利大放送
翌圣为您提供MolPure®磁珠法残留 DNA 样本前处理试剂盒(货号18461ES25)和MycAway™支原体qPCR检测试剂盒(探针法)试用装,每个客户限申请1套(即2种试剂盒各1个),总量限购40个(最终解释权归翌圣生物所有)。
扫码填写您的试用申请




