
NGS mRNA建库完全攻略—从mRNA纯化到注意事项
mRNA测序简述
RNA高通量测序(RNA-sequencing,缩写为RNA-seq)是目前高通量测序技术中应用广泛的技术之一,它可以帮助我们了解各种比较条件下基因表达情况的差异。在所有差异类型的检测中,常见的方法为检测mRNA表达量的差异,而mRNA样本在上机测序之前会做怎么的处理?它与DNA样本的建库处理又有哪些不同?今天小翌跟大家详细讲解。
mRNA建库步骤
在上机测序之前,提取到的核酸样本都需要做处理,样本处理的过程就叫做建库,我们都知道DNA建库的本质是在核酸片段两端加上接头的过程,RNA建库的本质也是如此。RNA建库由于增加了RNA富集、片段化和反转成双链DNA几个步骤显得比DNA文库构建要繁琐些,下面我们一起看看 mRNA建库中的三个关键步骤:mRNA纯化、片段化处理和反转成双链DNA。
首先说到mRNA纯化,为什么要做mRNA纯化呢,这是因为通常抽提到的总RNA中,绝大部分都是核糖体RNA(rRNA),如图1所示:

图1.人类组织或细胞总RNA中各种RNA分布饼状图
核糖体RNA占90%以上,mRNA占2%~3%,剩下的为LncRNA、tRNA、miRNA等。
如果我们把所有的RNA都拿来测序,测到的绝大部分的数据都是rRNA,而rRNA在不同组织、器官当中的表达极度稳定,也就是说,这样的数据,并不能为实验者提供有用的信息,所以在很多的转录组测序实验中都选择纯化mRNA的方法来提高最终测序数据利用率。
那么,该怎样从总的RNA中,单独将我们想要的mRNA纯化出来呢 ?
mRNA纯化的方法主要包括两种:poly(A)纯化法和去除rRNA法
poly(A)纯化法
Poly(A)纯化法主要应用在真核生物的mRNA纯化,大部分真核生物的mRNA结构与原核生物的mRNA结构有明显区别,真核生物的mRNA的3’端具有poly(A)尾结构,因此可以选用Oligo(dT)磁珠直接进行靶向杂交富集(见图2);当然,也可以通过使用Oligo(dT)引物进行反转录以扩增捕获带有poly(A)的mRNA,但是引物捕获由于特异性差和富集度低,因此在实际操作中还是以磁珠纯化的方法较多。
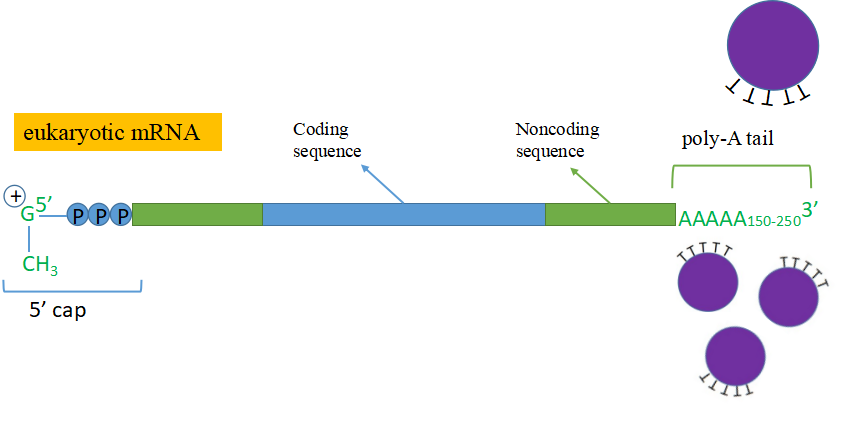
图2. 利用Oligo(dT)磁珠特异性吸附真核生物mRNA示意图
注意:利用Oligo dT磁珠对mRNA进行富集和纯化时,在移除上清时要等磁珠被彻底吸附后(上清澄清)在磁力架上小心进行,避免吸到磁珠,以免造成mRNA的损失。加入片段化试剂前,要将Bead Washing Buffer吸干净,否则会影响片段化结果,可以在用大规格枪移除液体之后再用10 uL的移液枪移除残留的Washing Buffer(或者直接用200 uL的黄枪头套着10 uL的白枪头使用)。
去除rRNA法
针对类似原核生物中没有poly(A)结构的mRNA和部分样品降解的总RNA样本(如FFPE样本),无法直接富集,只能通过去除rRNA的方法来达到富集mRNA的目的。
目前所涉及的去除rRNA方法主要如下:
1. 探针杂交捕获法(Ribo-Zero-seq)
原理:使用特异性的DNA探针或RNA探针与rRNA杂交,然后利用链霉亲和素磁珠吸附去除rRNA。
优点:高效、特异性好,适用于多种物种的rRNA去除。
缺点:需要根据研究物种的rRNA序列设计特异性的杂交探针。
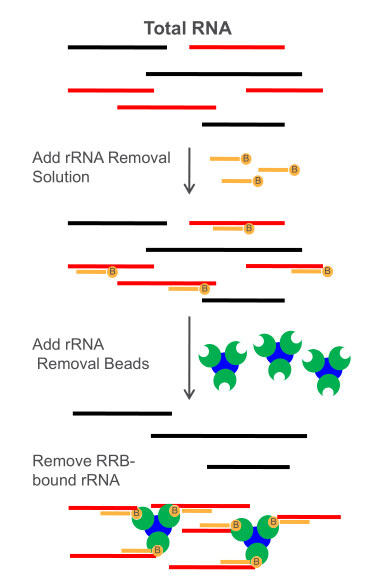
图3. 利用探针杂交捕获法(Ribo-Zero-seq)去除rRNA示意图
2. RNase H介导的消化法
原理:利用特异性的DNA探针与rRNA杂交,RNase H能够特异性消化DNA-RNA杂交链中的RNA单链,而未与探针杂交的mRNA、lncRNA等不受影响,从而富集目标RNA。
优点:高效、特异性好,适用于多种物种的rRNA去除。
缺点:需要设计特异性的DNA探针。
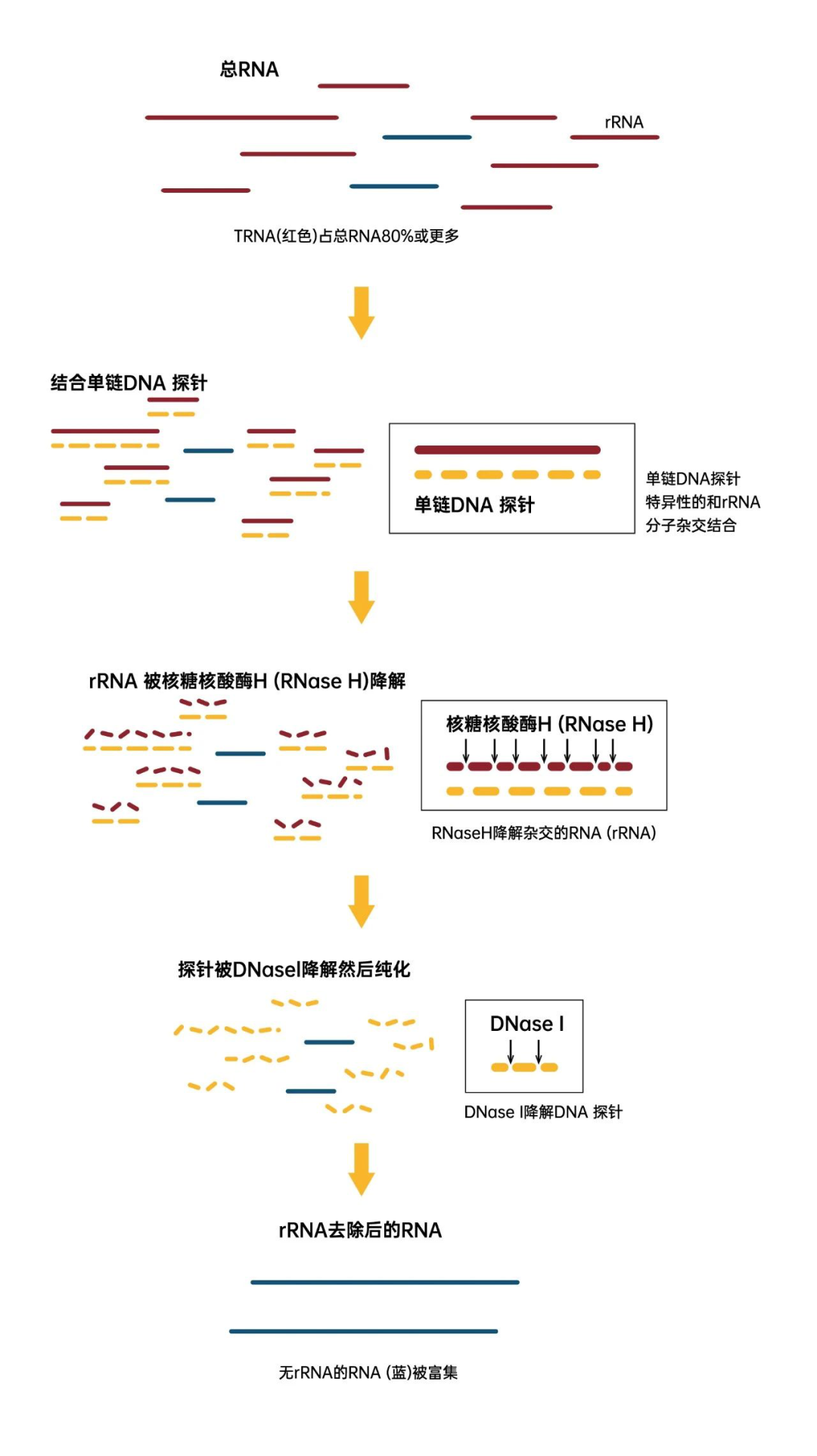
图4. RNase H介导的消化法去除rRNA示意图
3. 一步法靶向rRNA快速去除技术
原理:利用强结合能力的逆转录阻碍探针优先封闭靶RNA,阻止逆转录酶通过逆转录阻碍探针锁定的靶RNA位点,从而达到在逆转录过程中去除靶RNA的目的。
优点:操作简单,仅需一步加样操作,耗时仅3分钟,无需额外的纯化操作,对非靶RNA保留完整、特异性强、效率高和基因检出数高。
缺点:需要特定的试剂盒。
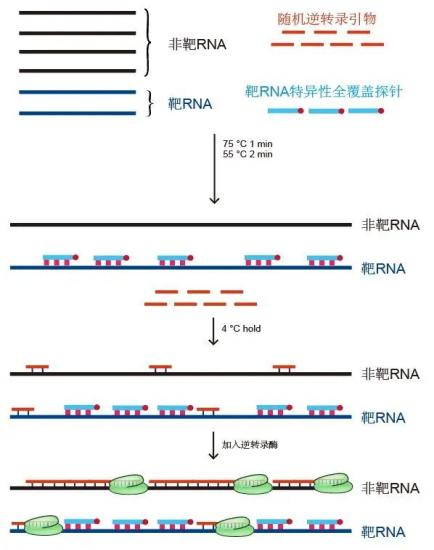
图5.一步法靶向rRNA快速去除
4、磁珠法rRNA去除技术
设计特异性的DNA探针,这些探针能够与需要去除的rRNA特异性杂交。探针通常带有生物素标记,以便与链霉亲和素磁珠结合。
优点:
-
高效:能够高效去除rRNA,保留目标RNA。
-
特异性高:通过特异性探针设计,减少非特异性结合,提高去除效率。
-
适用范围广:适用于多种物种的rRNA去除,包括原核生物和真核生物。
缺点:成本较高
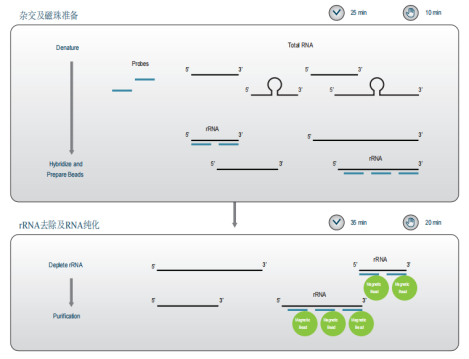
图6.磁珠法rRNA去除原理图
mRNA纯化之后的文库构建通常有两种思路,一种是先用oligo(dT)引物反转录mRNA,再进行cDNA的片段化,另外一种则是先将mRNA打断,再结合随机引物进行反转录。
1)先用oligo(dT)引物反转录mRNA,再进行cDNA的片段化:此方法先得到长片段的cDNA,反转成双链cDNA之后再利用DNA片段化的方法进行片段化(酶切法或机械法),得到片段化dsDNA之后的建库方法与DNA建库方法完全一致。
2)先将mRNA打断,再结合随机引物进行反转录:此方法中的mRNA打断方法包括碱处理法、金属离子(Mg2+、Zn2+)溶液处理法、酶(RNaseIII)处理法,mRNA片段化之后应立即进行一链cDNA合成,因为mRNA在该体系下非常容易降解。选择金属离子(Mg2+、Zn2+)溶液处理法打断时需根据需要的文库片段大小选择合适的打断温度和时间。(一般设置为150-200bp 94°C,15 min;200-300bp 94°C,10 min;250-550 bp 94°C,5 min)
两种片段化结果,对后期的转录本有不同的偏好性,对应数据可参考下图:
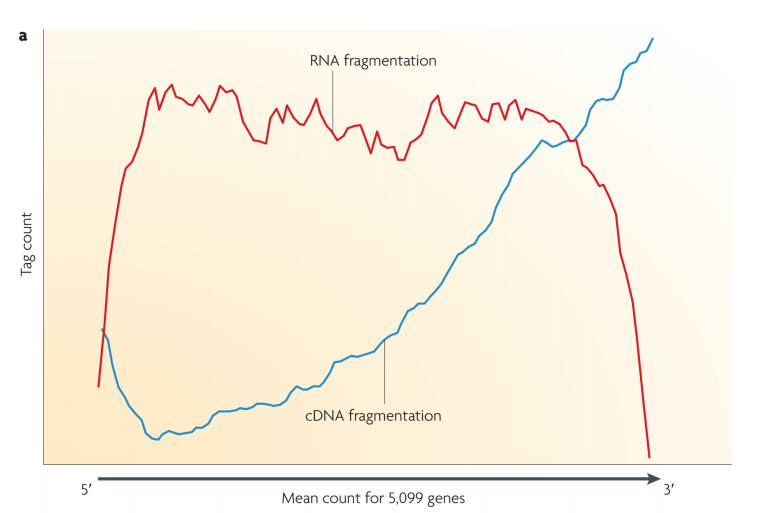
图7.不同方法建库对后期读数的偏好性,红线代表RNA片段化,绿线代表反转为cDNA后片段化
先针对mRNA进行打断再进行反转录获得测序reads主要是针对基因本体的;若先反转录,尤其是结合oligo(dT)进行反转录获得的测reads对转录本3'端具有比较强的偏好性,所以在mRNA-Seq中建议采用先对mRNA打断再进行反转录的文库构建方法。
在做mRNA测序文库构建的时候,一般分为常规建库和mRNA链特异性建库。链特异性文库和常规mRNA文库的区别在于合成第二链cDNA时加入的核苷酸种类,如果要构建常规mRNA文库,则加入dNTP,而如果要构建链特异性mRNA文库则加入dNTP中使用dUTP代替dTTP,这样合成的第二链cDNA含有U碱基,后期再通过识别U碱基的方法去除第二链cDNA,从而达到构建链特异性mRNA的效果,具体方法包括:使用特殊酶不扩增带有U碱基的链;使用UDG酶去除带有U碱基的合成链。
注意事项
小翌总结了mRNA建库中的几个小Tips,希望大家在mRNA建库的时候可以少走一些弯路:
Tip1:mRNA建库方法对RNA的完整度有较高的要求。也就是说,只有在mRNA大部分是完整的状态下,才能得到比较好的效果。
Tip2:尤其是选择磁珠吸附的方法,我们知道磁珠它所吸附的是Poly(A)的那些序列。那么如果mRNA发生了降解,也就是mRNA断掉了,那么磁珠所吸附下来的片段,都是那些靠近3'端的那些断片,而那些5'端的断片呢,是吸附不下来的。会在富集过程中被洗脱掉,如图8所示,那么,接下来的数据分析当中,就会发生一定的数据偏差。
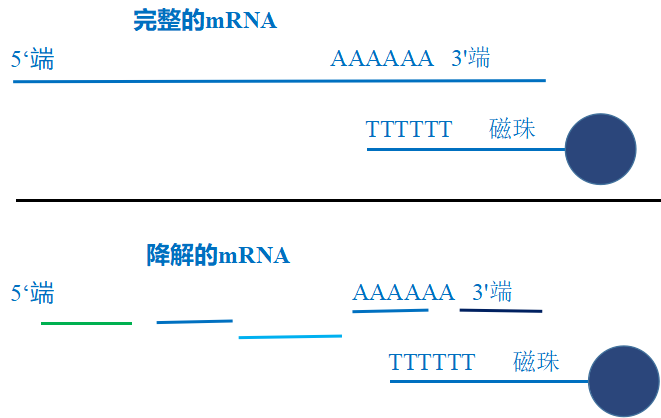
图8. 磁珠吸附正常的mRNA和磁珠吸附降解的mRNA示意图
如何保证能够测到尽可能完整的mRNA序列呢?
建库之前先对总RNA进行一次质量检测,一般是用Agilent公司出品的Bioanalyzer 2100毛细管电泳仪,对总RNA样本进行一次电泳质检。Bioanalyzer会根据18S和28S这两个核糖体RNA的电泳峰是否高、是否尖,来判断RNA的质量,并且会给出一个分值。这两个峰越高、越尖,也就说明RNA的降解就越少,完整度就越高。相对应的分值也会越高。反之,打分就会低。这个分值就是我们常说的“RIN”值。RIN值最高是10分,最低是0分。推荐大家使用RIN值在8.0以上的RNA进行建库和测序。
以上就是小翌为大家解析的mRNA建库的关键步骤,此外,小翌还专程为大家绘制了一幅mRNA建库的标准流程图,希望可以帮助大家对建库的整套流程有更好的认识。
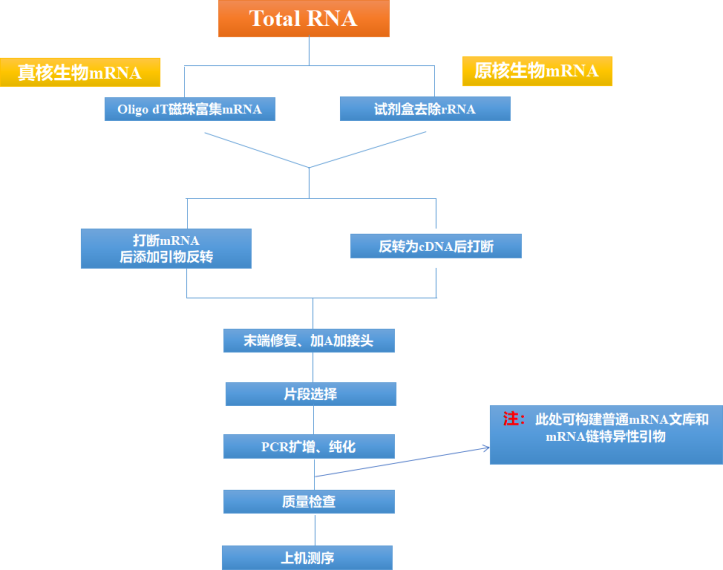
图9. mRNA建库流程图
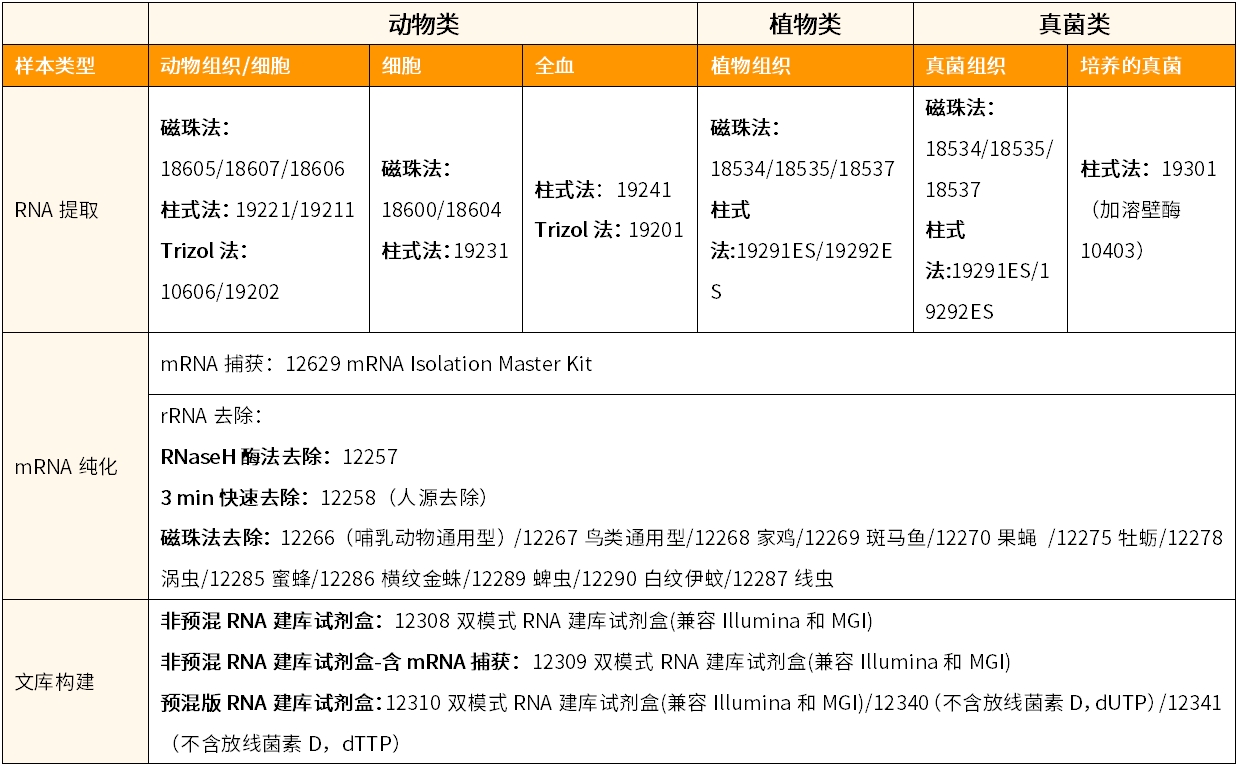
翌圣mRNA文库构建解决方案
推荐商品


