
过瘾!一文搞懂qPCR引物设计,实验达人必备技能!
不知各位小伙伴在做荧光定量qPCR实验的时候有没有遇到过以下这些情况?
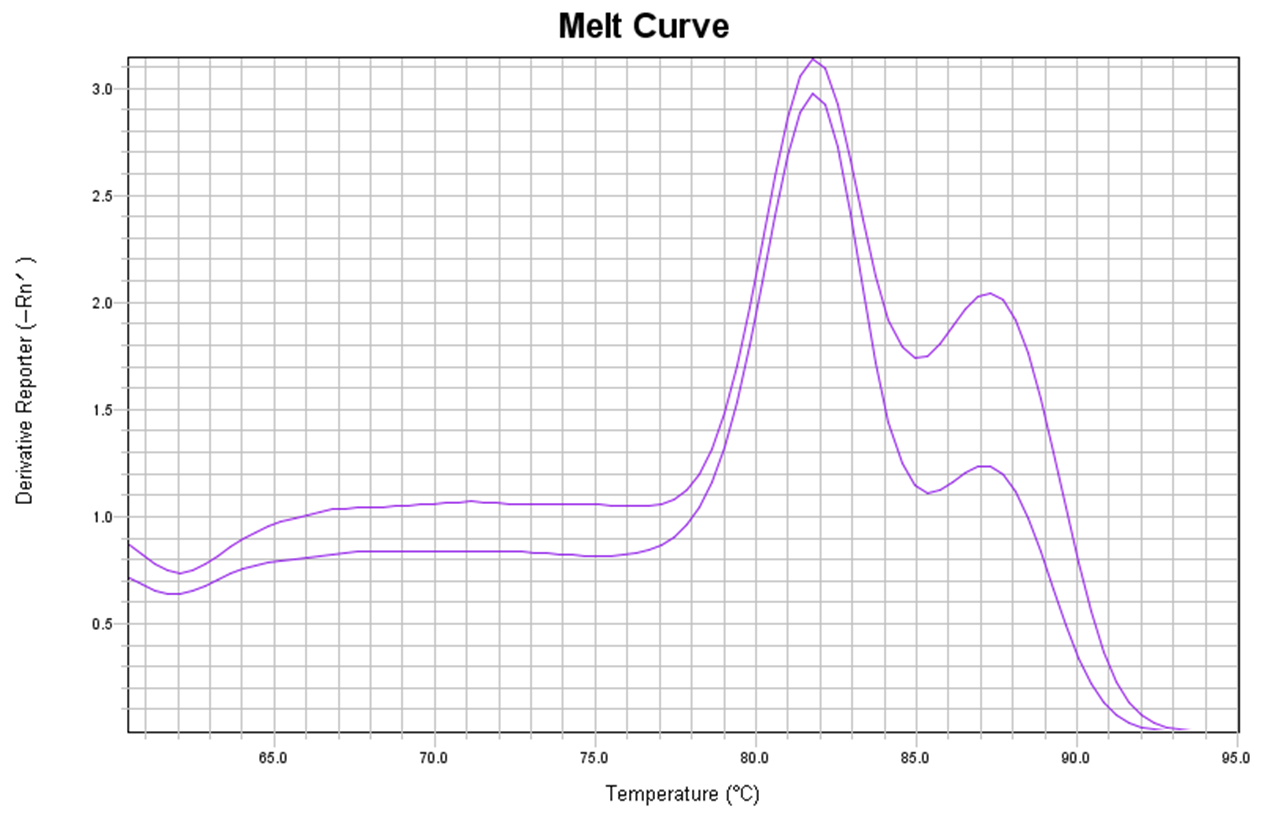
主峰左边有杂峰,疑似引物二聚体
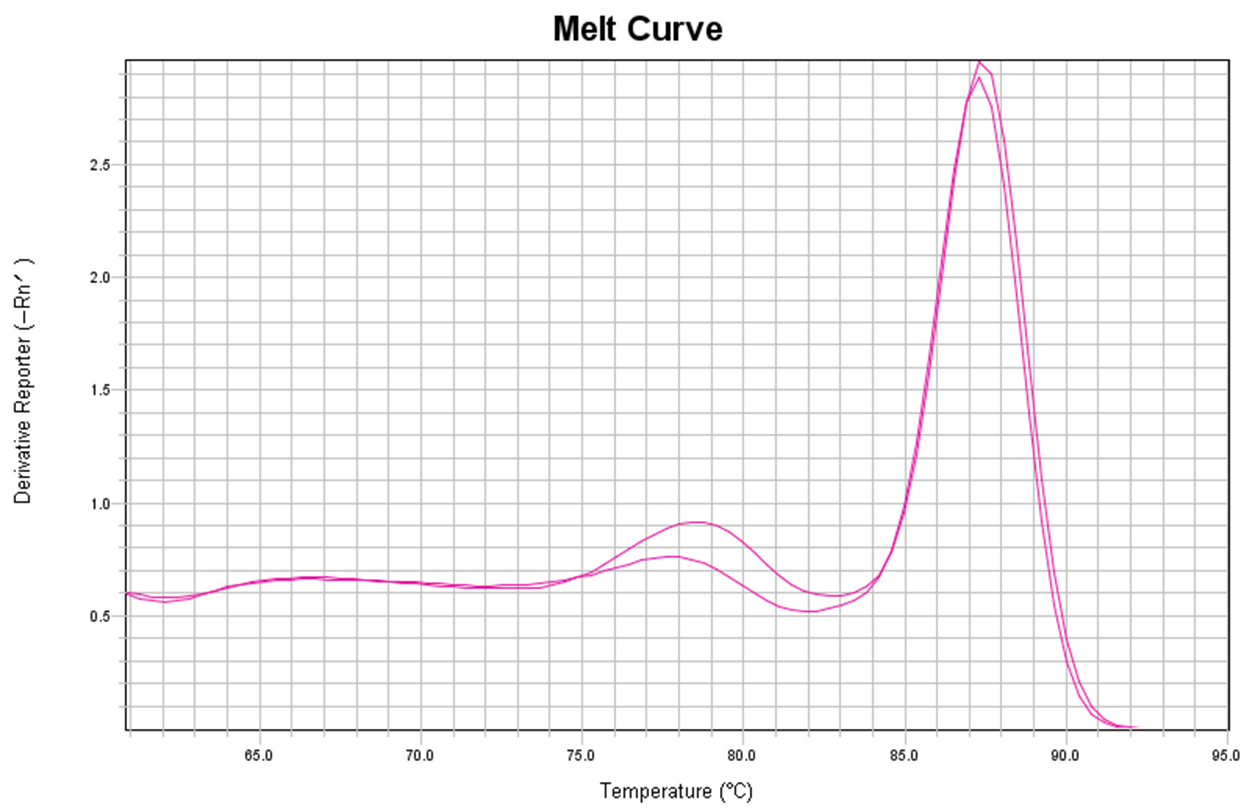
主峰右边有杂峰,疑似非特异扩增
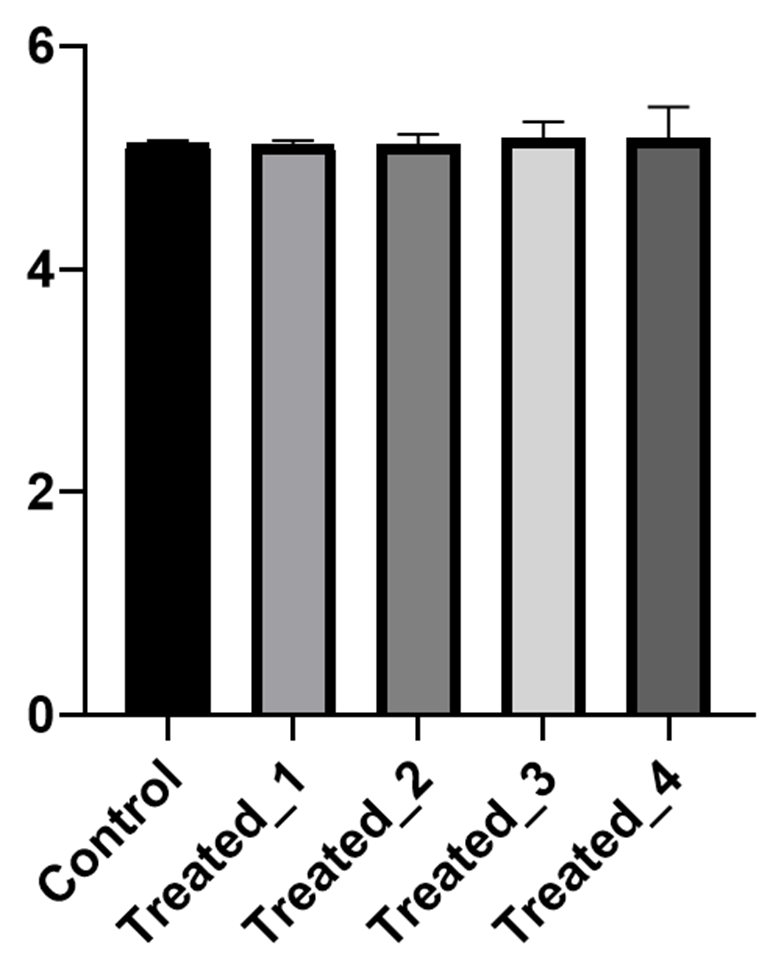
基因表达无差异
每当出现这些情况,就意味着可能要重新做实验。为确保下一次实验不出错,了解其原因十分重要,可能是体系污染导致,也可能是扩增特异性不足导致。在这里小翌教大家从引物设计的角度提升扩增特异性!
在进行qPCR引物设计时,相信有不少小伙伴听过类似“跨内含子设计”的要求,那么,要跨内含子设计是啥?为什么要跨内含子设计呢?
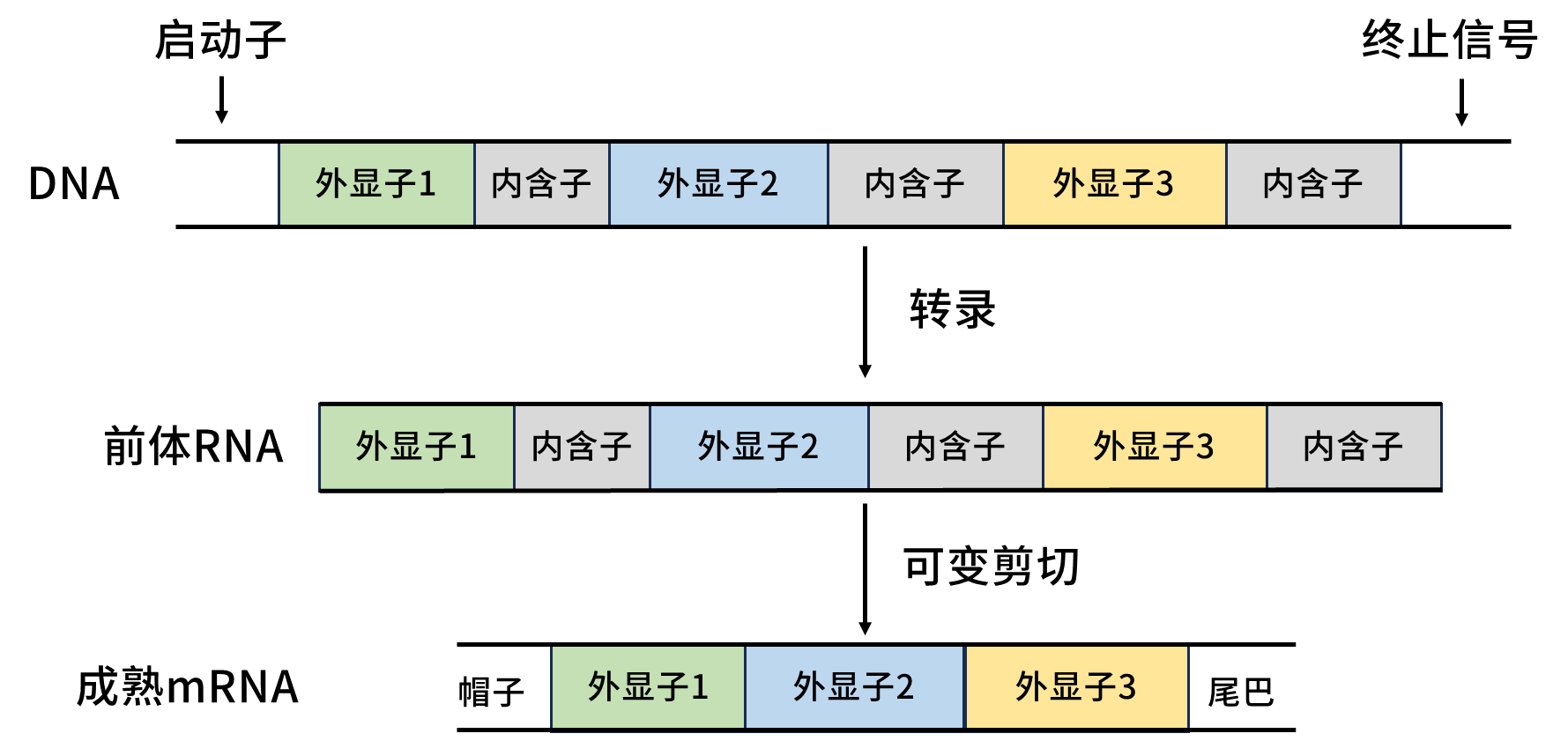
以人基因组来说,平均每个基因有8.8个外显子和7.8个内含子,80%的外显子长度小于200bp,少于10%的内含子长度在1100bp以上,不到0.01%的内含子长度小于20bp。
我们在进行一些荧光定量qPCR实验之前,需要提取RNA和反转录合成cDNA,cDNA作为模板用于分析基因表达情况。故在DNA序列上设计引物需避开内含子序列区域,选用外显子区域序列用于引物设计。看到这,有小伙伴会有疑问,那为啥不叫“外显子设计”或者“去内含子设计”呢,为啥要称为“跨内含子设计”?
是的,引物还需要跨内含子,在设计引物时需要让一端引物或者两端引物跨越内含子,否则荧光定量qPCR的熔解曲线可能出现双峰或者多峰。
在单个外显子上进行上下游引物设计
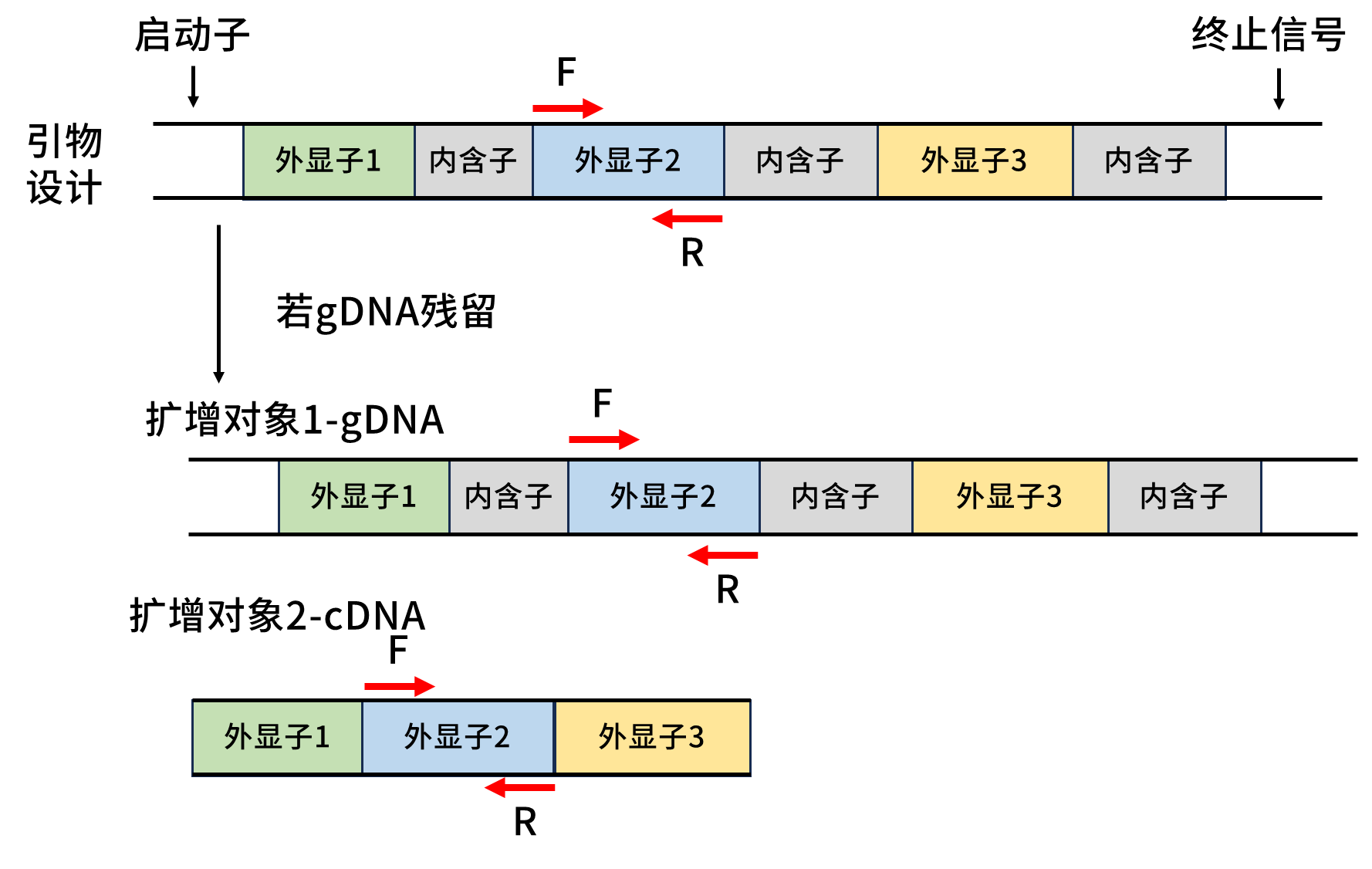
在这种情况下,由于产物序列一致且大小一致,从熔解曲线上难以分辨,看上去都是单一的熔解峰,但是在进行数据分析时,往往会有一种感觉,就是基因表达无差异或者差异结果不稳定。
在两个外显子上进行上下游引物设计
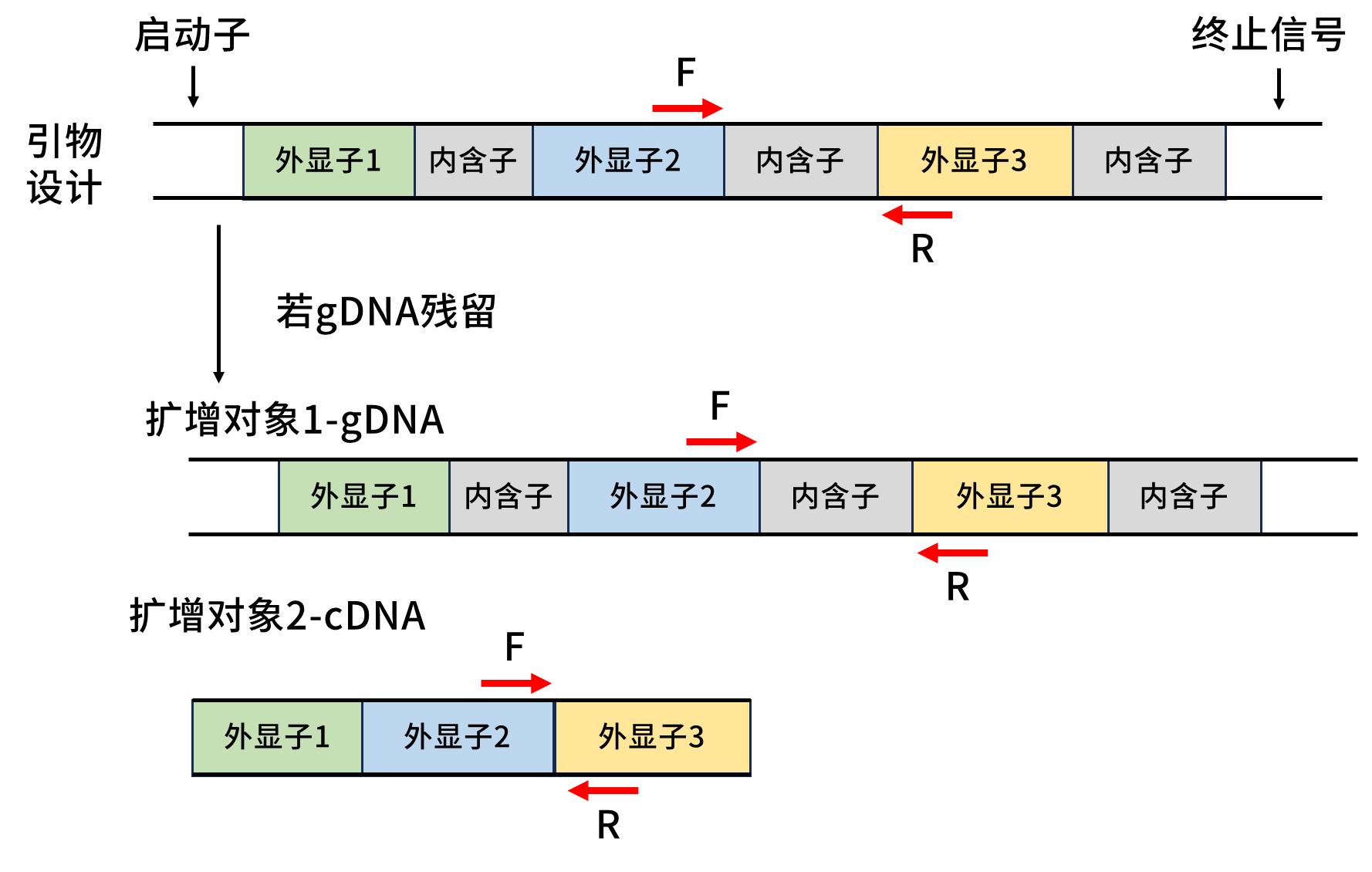
在这种情况下,产物差距就比较大了,基因组DNA(gDNA)扩增产物较cDNA扩增产物更长,熔解温度(Tm值)更大,在熔解曲线上表现出来则是主峰右侧的杂峰。当然还有一种情况,当以gDNA为模板所需扩增的片段很长,难以扩增,最终产物相对单一,熔解曲线峰也是相对单一的。
建议的上下游引物设计方式
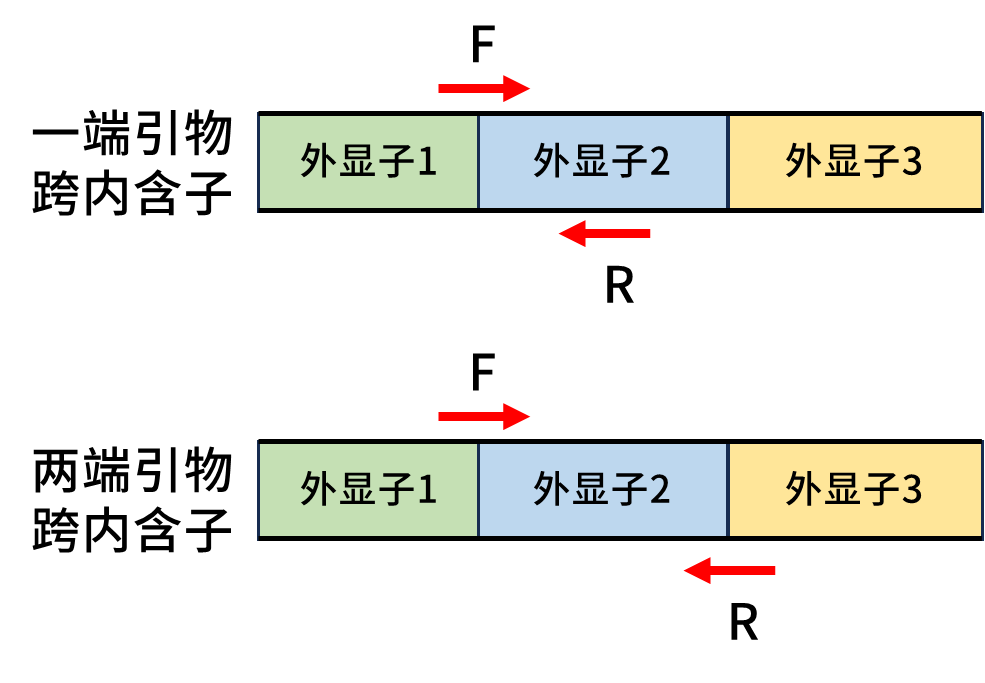
引物跨内含子设计后,对于引物序列本身还有什么要求呢?接下来小翌给大家分享引物设计原则~
qPCR引物设计原则
|
参考项 |
标准参考 |
|
产物长度 |
80-300bp |
|
引物长度 |
17-25 base |
|
GC含量 |
40-60%(45-55%为最佳) |
|
Tm值 |
上游引物F和下游引物R的Tm值不能相差太大,最好不超过1℃。 |
|
引物序列 |
A、T、C、G整体分布尽量均匀。避开T/C或A/G的连续结构。 |
|
3末端序列 |
避免GC rich或AT rich。3端碱基最好为G或C,尽量避免末端碱基为T。 |
|
互补序列 |
避开引物内部或两条引物之间有3个碱基以上的互补序列。 两条引物间的3末端避开有2个碱基以上的互补序列。 |
|
特异性 |
使用BLAST检索确认引物的特异性。 |
产物长度
首选的产物扩增长度为80-200bp,必要时可选择加长。通常扩增效率随着扩增产物长度增大而减小。但是不能太短,否则难以区分是引物二聚体还是扩增产物。
末端序列
引物末端最好是G或C,因为GC碱基之间是三个氢键连接,能够保证引物和模板连接的稳定性。
互补序列
引物3端通常是DNA聚合酶延伸的起点,如果两条引物在3端互补性过高,则更易形成引物二聚体,从而降低PCR的扩增效率和特异性。
若引物自身的互补性高,则会发生自身退火,形成发夹结构,或者两条一样的引物形成二聚体,从而降低PCR的扩增效率和特异性。
聊完了原则上qPCR引物如何设计之后,接下来让我们进入小伙伴们最为关注的实战操作。在这里小翌给大家介绍三种方式设计qPCR引物!
Primer3Plus
网址链接:https://www.primer3plus.com
Load server settings 选择qPCR,后上传设计引物的序列或粘贴需要设计引物的序列。按照自己的引物设计需求,选择使用排除的区域Excluded Regions、目的扩增区域Targets和包含区域Included Region。
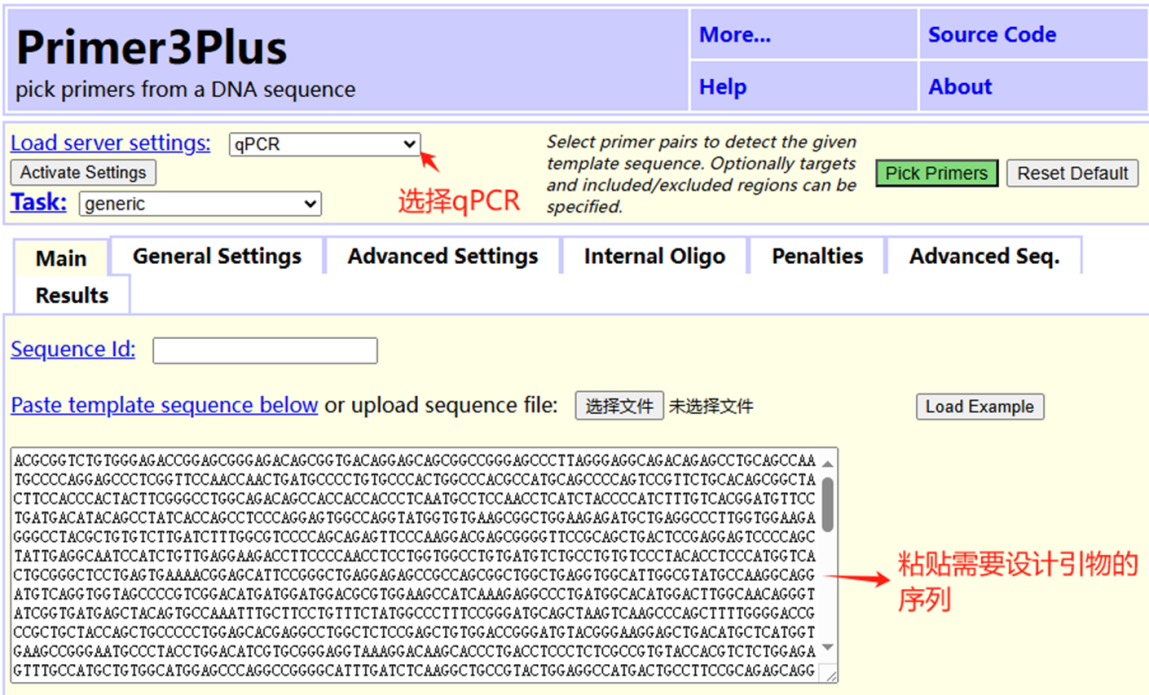
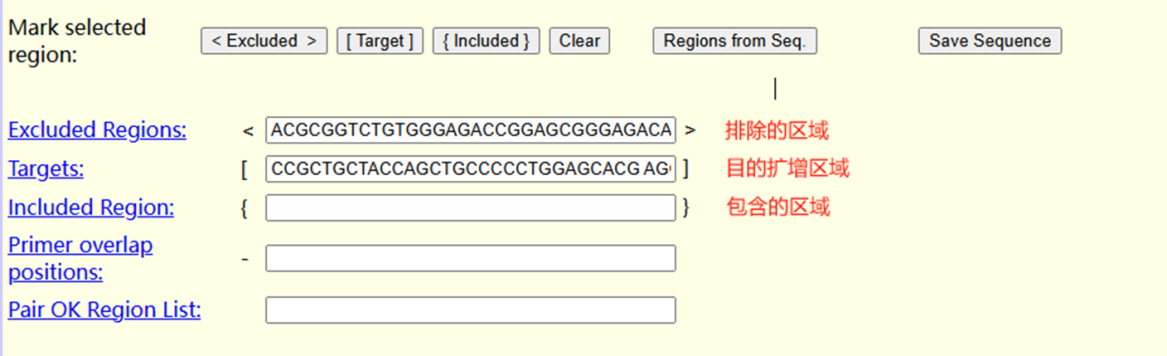
点击General Settings按照荧光定量qPCR引物设计原则进行相关参数设置,例如扩增产物长度Primer Size Ranges、引物长度Primer Size、Tm值Primer Tm、GC含量Primer GC%。完成相关参数设置后,点击右上方绿色按钮Pick Primers,即可获得相关引物。

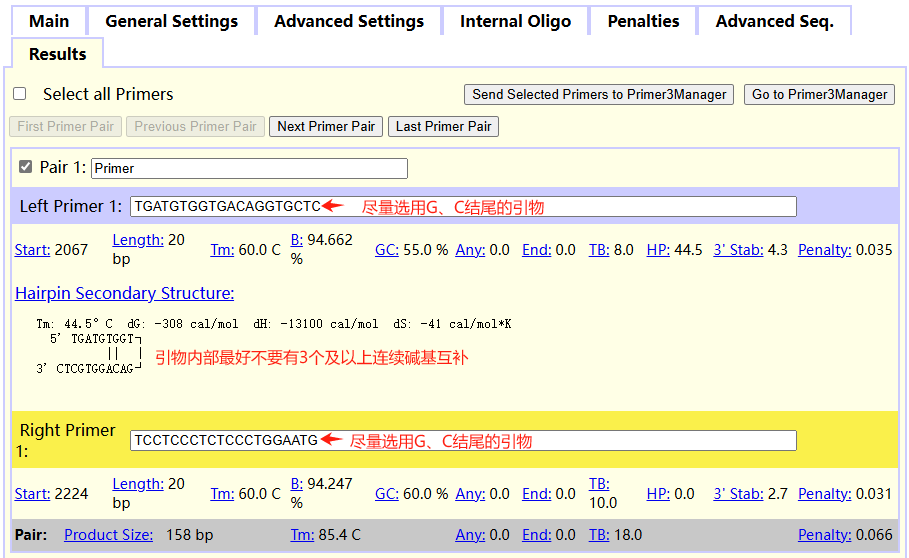
获得相关引物序列后,到NCBI blast ( https://www.ncbi.nlm.nih.gov/tools/primer-blast/)进行引物特异性验证。
Primerbank
网址链接:https://pga.mgh.harvard.edu/primerbank/
它是哈佛大学的一个PCR引物数据库,包含 306,800 多种引物,涵盖大多数已知的人类和小鼠基因。通过 Real Time PCR 测试了与 27681 个小鼠基因相对应的 26855 对引物,然后对 PCR 产物进行琼脂糖凝胶电泳和测序。根据琼脂糖凝胶电泳结果,设计成功率为 82.6%(22187 对引物)。
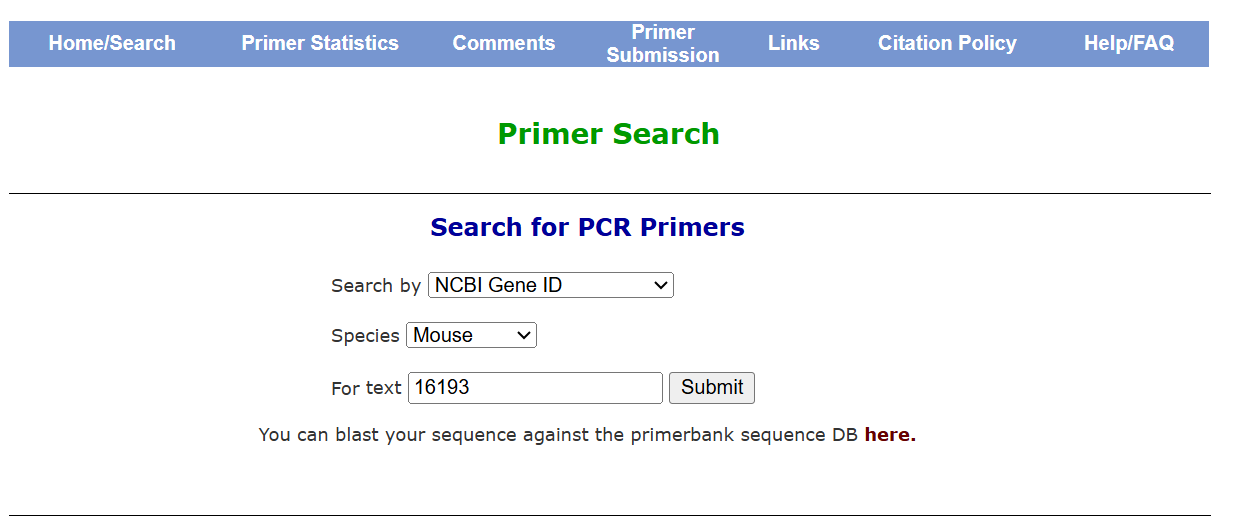
首先在NCBI上找到目的基因的gene ID,例如,这里选用老鼠基因IL6的gene ID 16193。

Search by选择 NCBI Gene ID,Species选择Mouse,输入16193,点击Submit,即可获得相关引物。
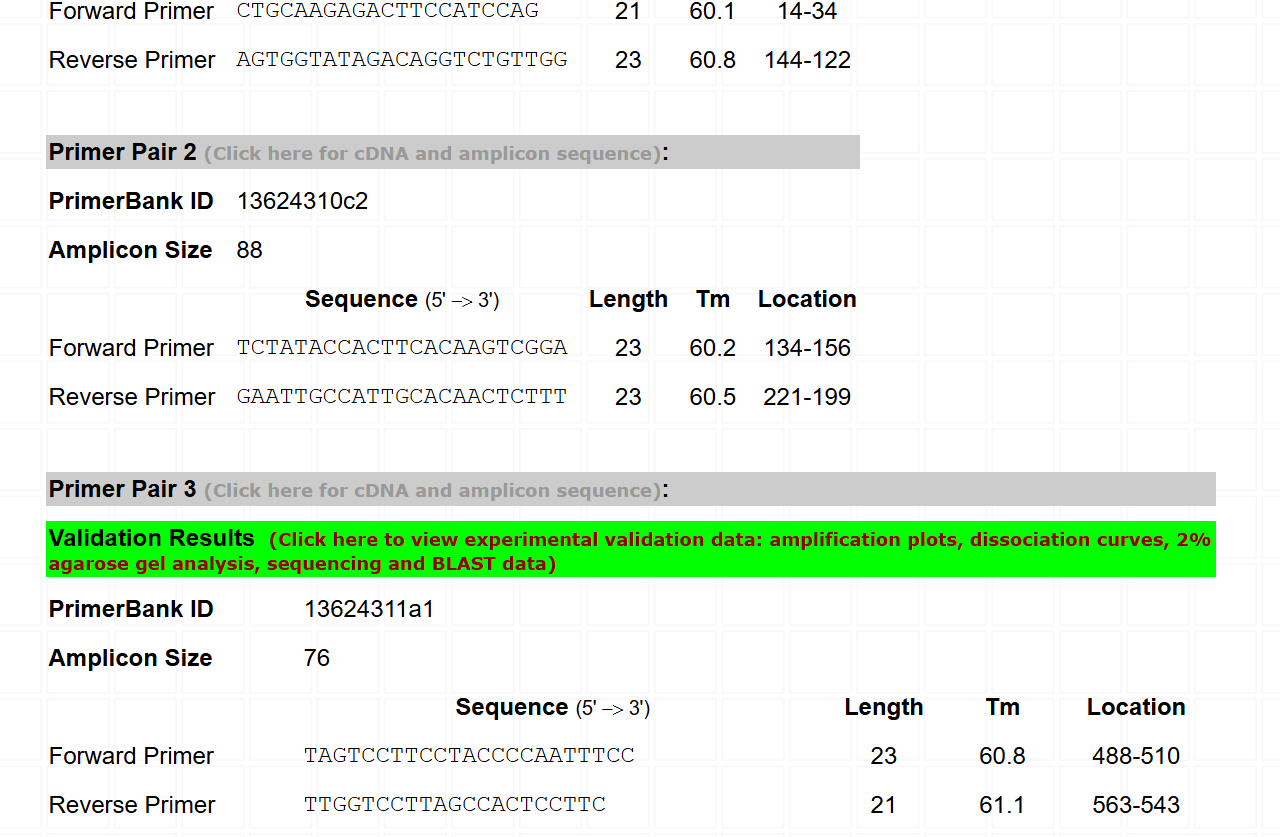
相关引物还有已验证的可视化结果。
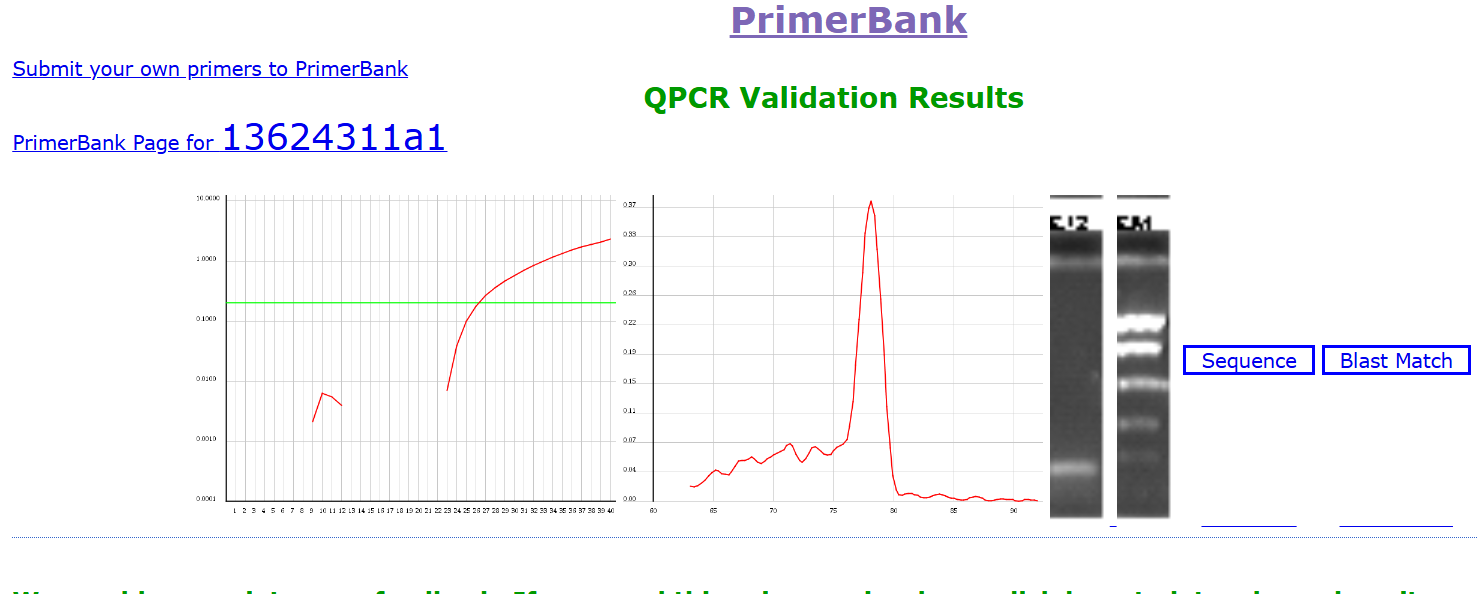
NCBI
网站链接:https://www.ncbi.nlm.nih.gov
打开NCBI,选择“Gene”,输入想要检测的基因名称,我们以人基因“ALAD”为例。
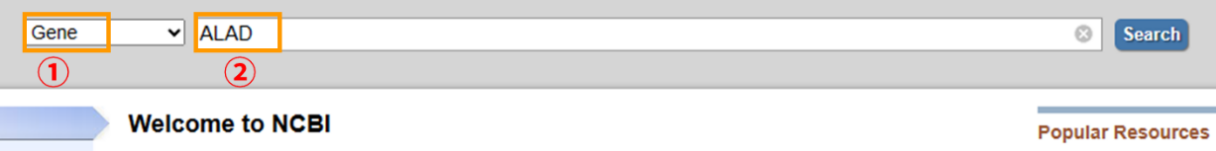
会出现很多搜索结果,我们选择需要的种属来源,即“human”,如果你知道Gene ID(每个基因有且只有一个特定的ID,类似于人的身份证),也可以根据Gene ID来选择。这里我们选择搜索结果中的第一个。
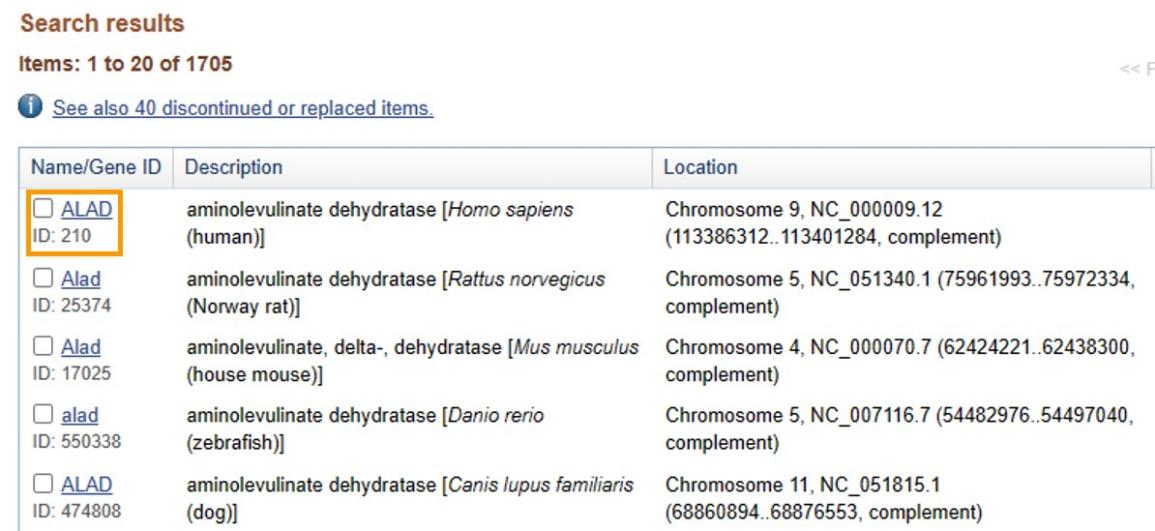
点开之后,会出现很长的页面,耐心往下拉,找到“mRNA and Protein(s)”栏下的NM编号,不同的NM编号,代表不同的isoform。选择想要检测的那一个。
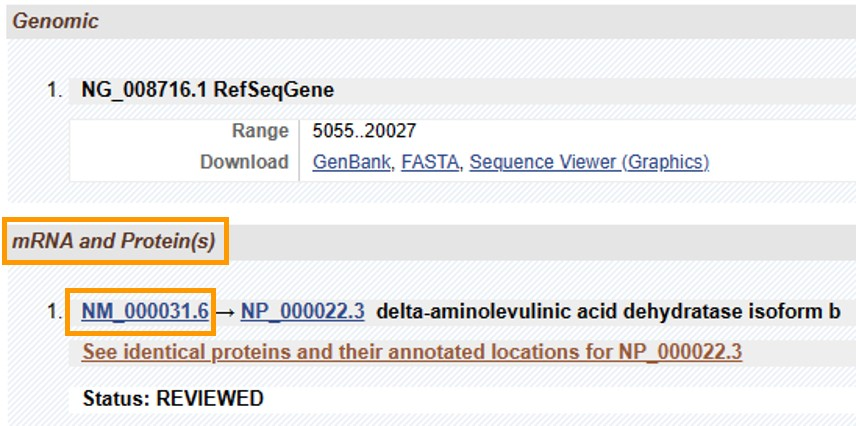
确定NM号点击后,可以看到这个isoform的一些基本信息。

例如:gene代表基因长度3129bp,CDS代表编码蛋白区域149-1141bp。
以及此基因的序列,如下图所示。此序列就是设计引物时对应的模板。

了解完我们所需的isoform后,我们可以点击右侧的“Pick Primers”,就能直接进入Primer-BLAST的页面进行引物设计了。

在弹出来的界面里粘贴模板序列或者输入基因序列号。另外需要设置“上下游引物位置”和“扩增子长度”。最主要的设置就是这两点了,另外还有“物种名称”、“数据库”等其他选项可以设置。
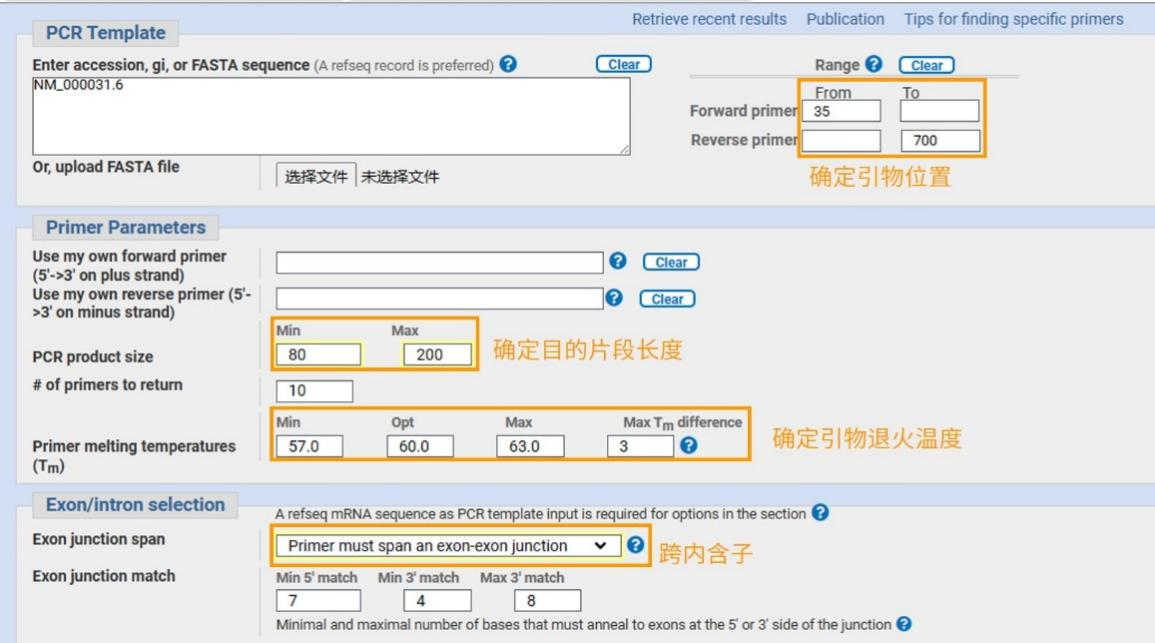
设置好之后,点击页面左下角的“Get Primers”。
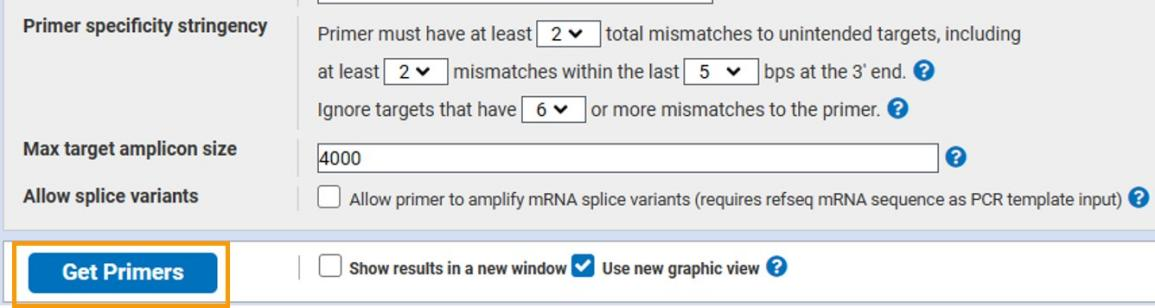
多对引物,选择合适的引物即可。

获得相关引物序列后,到NCBI blast ( https://www.ncbi.nlm.nih.gov/tools/primer-blast/)进行引物特异性验证。
新品预告

翌圣生物基因研究完整解决方案
产品特别推荐
|
方法 |
分类 |
产品名称 |
货号 |
|
RNA提取 |
同Trizol提取 |
TRIeasy™ Total RNA Extraction Reagent |
|
|
免氯仿升级版 |
TRIeasy™ Total RNA Extraction Reagent(Tcm Free) |
||
|
动物组织/细胞总RNA提取,避开有毒试剂,最快15 min完成 |
MolPure® Cell/Tissue Total RNA Kit细胞/组织总RNA提取试剂盒 |
||
|
反转录试剂 |
5 min一步gDNA去除&反转录预混液(下游应用qPCR) |
Hifair® AdvanceFast One-step RT-gDNA Digestion SuperMix for qPCR |
|
|
5 min快速反转,最长可满足14 kb cDNA合成,含gDNA去除(下游应用PCR/qPCR) |
Hifair® AdvanceFast 1st Strand cDNA Synthesis Kit |
11149/11150ES |
|
|
高质量第一链cDNA合成预混液,含gDNA去除(下游应用qPCR) |
Hifair® III 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) |
||
|
qPCR染料法 |
高特异高荧光值定量预混液(染料法) |
Hieff UNICON® Advanced qPCR SYBR Master Mix |
|
|
高灵敏通用型定量预混液(染料法) |
Hieff UNICON® Universal Blue qPCR SYBR Master Mix |
||
|
超高性价比定量预混液 (染料法),已发文章累计IF达到5000+ |
Hieff® qPCR SYBR Green Master Mix (No Rox) |
||
|
Hieff® qPCR SYBR Green Master Mix (Low Rox) |
|||
|
Hieff® qPCR SYBR Green Master Mix (High Rox) |

