
干货!常用PCR技术及原理
PCR(Polymerase Chain Reaction)即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,体系中加入dNTP、Mg2+以及延伸因子、扩增增强因子,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程,能快速特异地在体外扩增任何目的DNA。
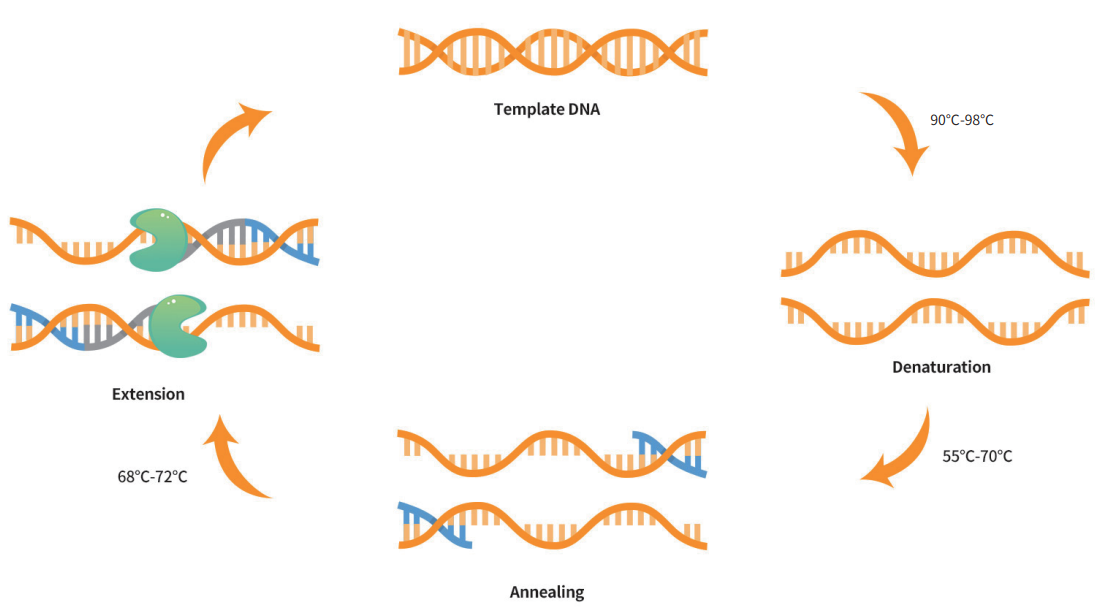
图1. PCR扩增流程
常规PCR的扩增开始时间并不是放进PCR仪,运转程序才开始扩增。当体系配置完成的时候,扩增就开始了,这可能会引起非特异性扩增出现,热启动PCR可以解决这一问题。什么是热启动PCR?当反应体系配制好后,在反应初始加热阶段或“热启动”阶段,酶修饰物在高温下(通常高于90 ℃)被释放,使得DNA聚合酶被激活。具体激活时间和温度取决于DNA聚合酶以及热启动修饰物的性质。该方法主要利用抗体、亲合配体、或化学修饰物等修饰物,来抑制的DNA聚合酶的活性。由于DNA聚合酶在常温下的活性被抑制,热启动技术为在常温下配制多个PCR反应体系提供了极大的便利,且无需牺牲PCR反应的特异性。
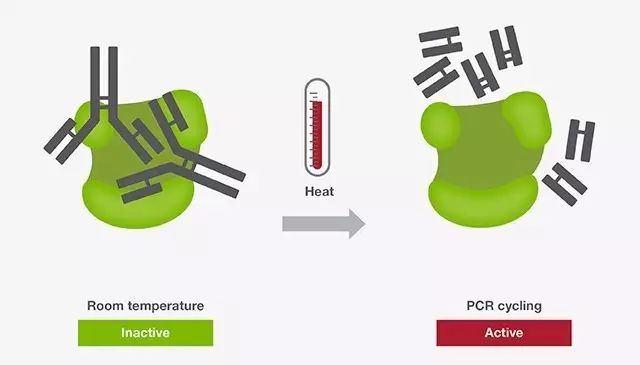
图2. 基于抗体热启动技术的DNA聚合酶
逆转录PCR(Reverse Transcription PCR,RT-PCR),是从mRNA逆转录成cDNA并以此为模板进行扩增的一种实验技术。实验流程是先提取组织或细胞中的总RNA,以Oligo(dT)为引物,利用逆转录酶合成cDNA,再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。
注:oligo(dT)是由数量少于20的脱氧胸苷酸连接而成的核苷酸链,它可以特异性地结合到mRNA的poly(A)尾端(多A尾),从而使逆转录酶只能逆转录含有poly(A)尾的mRNA。
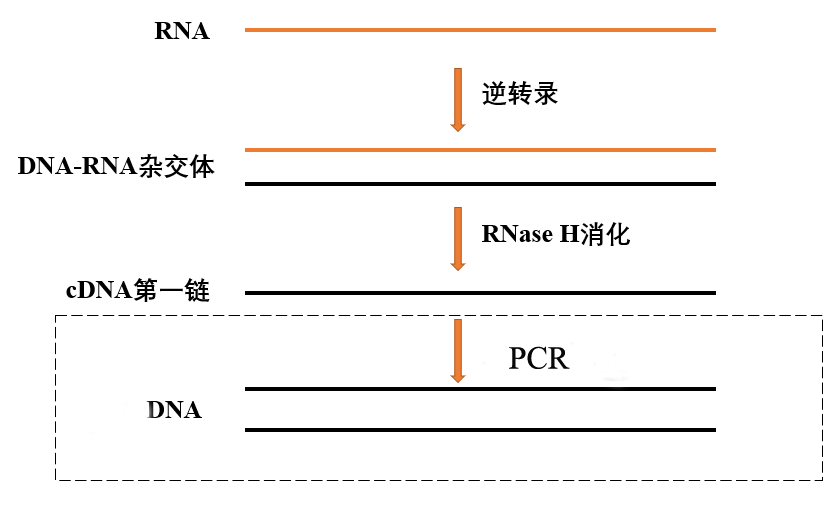
图3. 逆转录PCR原理
荧光定量PCR(Real-time Quantitative PCR,RT-qPCR)是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对模板进行定量分析的方法。常用的qPCR方法有荧光染料法(SYBR Green I)和探针法(TaqMan)。染料法(常用SYBR Green Ⅰ):在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,没有掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。探针法(常用TaqMan探针):探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物的形成完全同步。
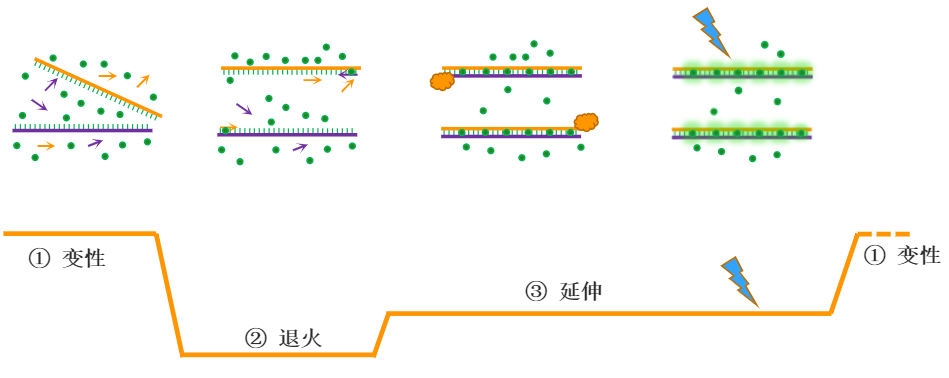
图4. 染料法原理
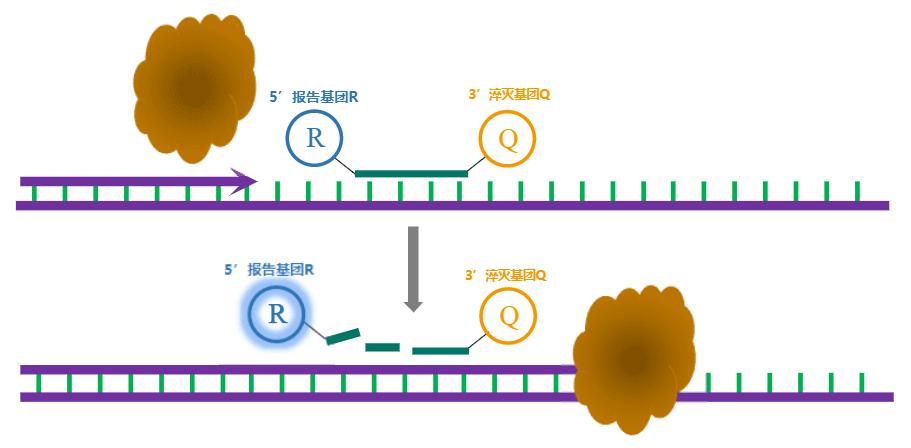
图5. 探针法原理
巢式PCR(Nested PCR)是指利用两套PCR引物进行两轮PCR扩增,第二轮的扩增产物才是目的基因片段。如果第一对引物(外引物)的错配导致非特异性产物被扩增,相同的非特异性区域被第二对引物识别并继续扩增的可能性非常小,所以通过第二对引物的扩增,PCR的特异性得到了提升。进行两轮PCR的一个优势在于:有助于从有限的起始DNA中扩增得到足量的产物。巢式PCR的实验原理为根据DNA模板序列设计两对引物,利用第一对引物(称为外引物)对靶DNA进行15-30个循环的标准扩增;第一轮扩增结束后将一小部分起始扩增产物稀释100-1000倍加入到第二轮扩增体系中作为模板,利用第二对引物(称为内引物或巢式引物,结合在第一轮PCR产物的内部,进行15-30个循环的扩增,第二轮PCR的扩增片段短于第一轮。
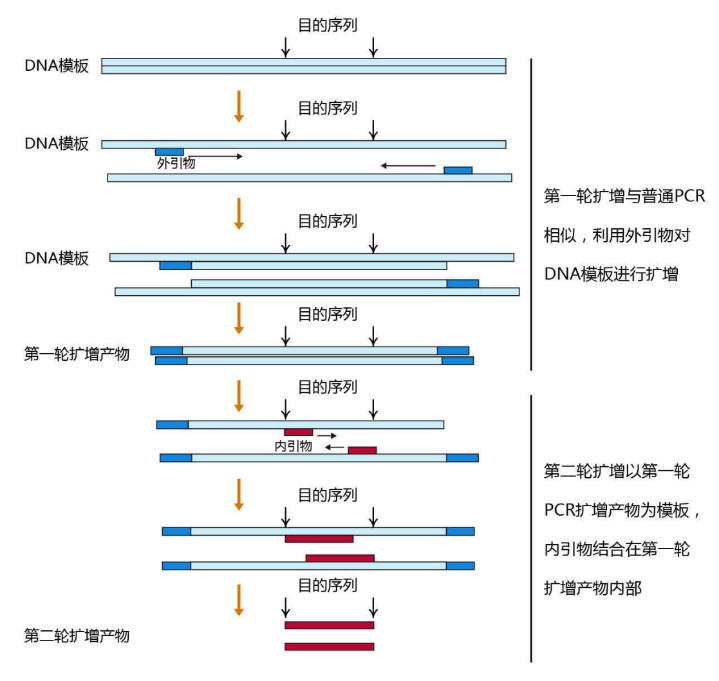
图6. 巢式PCR原理
降落PCR(Touchdown PCR)是一种通过调整PCR循环参数,提升PCR反应特异性的方法。在降落PCR中,前几个循环的退火温度设定为,比引物的最高退火温度(Tm)再高几度。较高的退火温度能有效减少非特异性扩增,但同时较高的退火温度会加剧引物与目标序列的分离,导致PCR的产量降低。因此在开始的几个循环中,退火温度通常设置为每个循环降低1℃,以增加体系中目的基因的含量。当退火温度降低到最佳温度时,剩余循环都维持此退火温度。通过这种方法,在PCR过程中,所期望的PCR产物得到有选择性的增加,而几乎不会出现非特异性的产物。
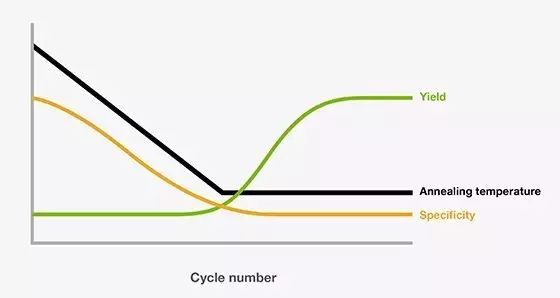
图7. 降落PCR
注:通过以较高的温度起始,再随着循环逐渐降低到最佳退火温度(黑色曲线),这种PCR方法可提升反应特异性(黄色曲线)。目标扩增子的产量(绿色曲线)相应的得到富集。
直接PCR是指直接从样品扩增目标DNA,无需进行核酸分离纯化。直接PCR分两类,直接法:取少量样本直接加入到PCR Master Mix中,进行PCR鉴定;裂解法:样本取样后,加入裂解液中,裂解释放基因组,取少量裂解上清液加入到PCR Master Mix中,进行PCR鉴定。这种方法简化了实验流程,减少了动手操作时间,同时可避免纯化步骤DNA的损失。
推荐具有高合成能力的DNA聚合酶用于直接PCR扩增。细胞碎片、蛋白、脂质和多糖也随DNA释放到裂解液中,它们会抑制PCR反应。具有高合成能力的DNA聚合酶可以耐受这些抑制剂,从而使直接PCR成为可能。具有高合成能力的聚合酶通常具有更高灵敏度,因此可从未纯化样本中扩增微量的DNA。
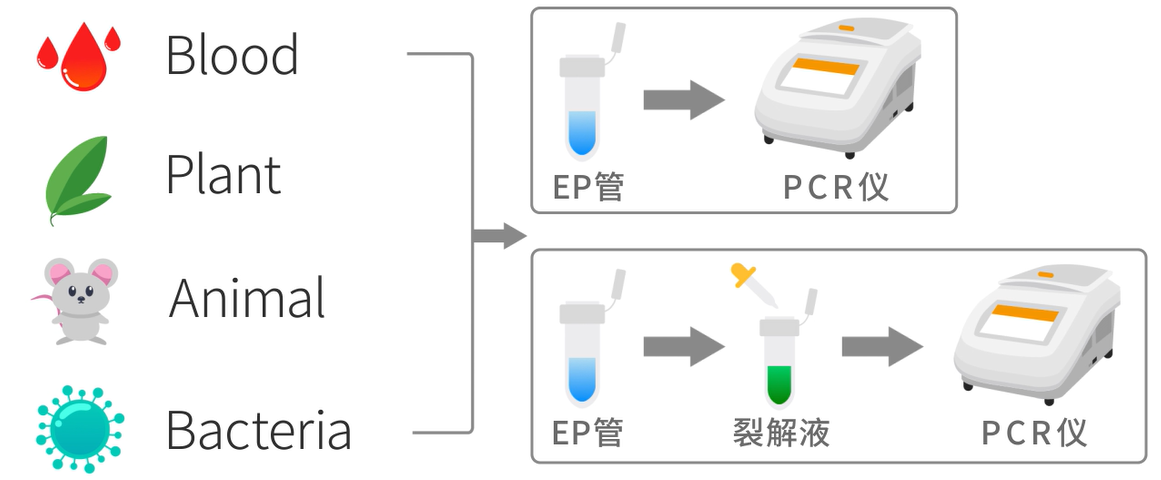
图8. 直接PCR示意图
重叠延伸PCR技术 (gene splicing by overlap extension PCR,SOE PCR),是采用具有互补末端的引物,使 PCR 产物形成了重叠链,从而在随后的扩增反应中通过重叠链的延伸,将不同来源的扩增片段重叠拼接起来的技术。这项技术目前主要有两个应用方向:构建融合基因;基因定点突变。
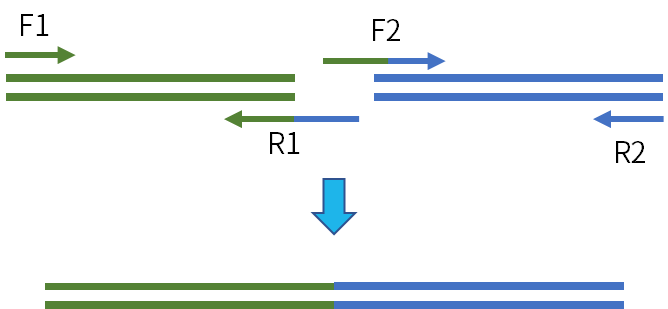
图9.重叠延伸PCR技术用于构建融合基因的原理
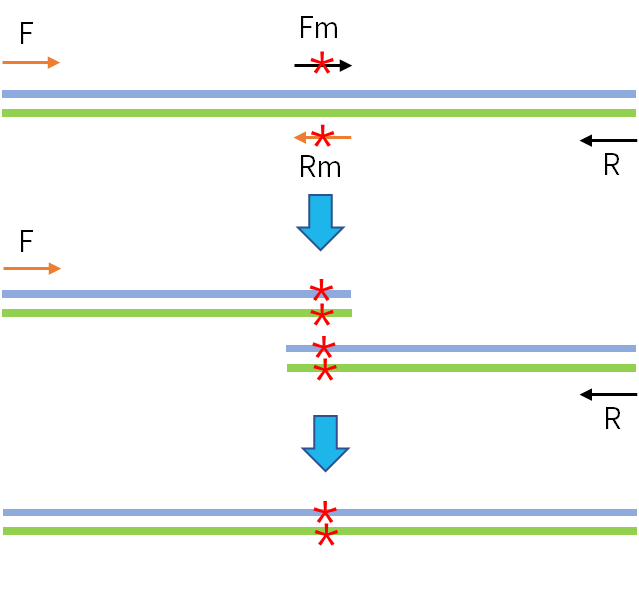
图10. 重叠延伸PCR技术用于基因定点突变的原理
反向PCR (Inverse PCR, IPCR)是用反向的互补引物来扩增两引物以外的DNA片段,对某个已知DNA片段两侧的末知序列进行扩增。反向PCR设计之初是为了用于确定邻近未知区域的序列,多用于研究基因的启动子序列;致癌性染色体重排,如基因融合、易位和转座;以及病毒基因整合,现在也常用于定点突变,复制一个具有预期突变的质粒。反向PCR流程,首先对模板进行限制性酶切消化和连接,选定的限制性内切酶不可剪切已知序列,从而使连接发生于侧翼未知序列之间。使用低浓度的酶切DNA片段优化连接步骤,使其倾向于自我连接而非多片段连接(即形成连环体)。完成自我连接后,从DNA的已知区域启动反向PCR。所获得的扩增子每个末端都含有部分已知DNA序列。随后,对这些扩增子进行测序,检测上述已知序列的相邻区域。
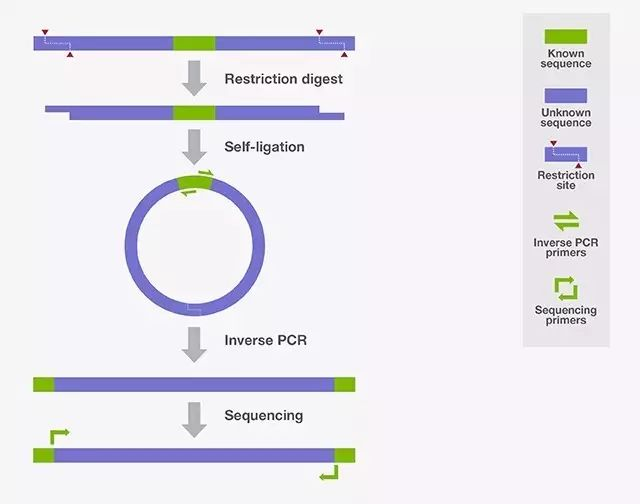
图11. 使用反向PCR来扩增和鉴定临近未知区域序列
数字PCR(Digital PCR,dPCR)是一种核酸分子绝对定量技术。当前核酸分子的定量有三种方法,光度法基于核酸分子的吸光度来定量;实时荧光定量PCR(Real Time PCR)基于Ct值,Ct值就是指可以检测到荧光值对应的循环数;数字PCR是最新的定量技术,基于单分子PCR方法来进行计数的核酸定量,是一种绝对定量的方法。主要采用当前分析化学热门研究领域的微流控或微滴化方法,将大量稀释后的核酸溶液分散至芯片的微反应器或微滴中,每个反应器的核酸模板数少于或者等于1个。这样经过PCR循环之后,有一个核酸分子模板的反应器就会给出荧光信号,没有模板的反应器就没有荧光信号。根据相对比例和反应器的体积,就可以推算出原始溶液的核酸浓度。除了基因表达和拷贝数检测,数字PCR也适用于诸如低频等位基因的辨别、病毒滴定和二代测序文库的绝对定量。
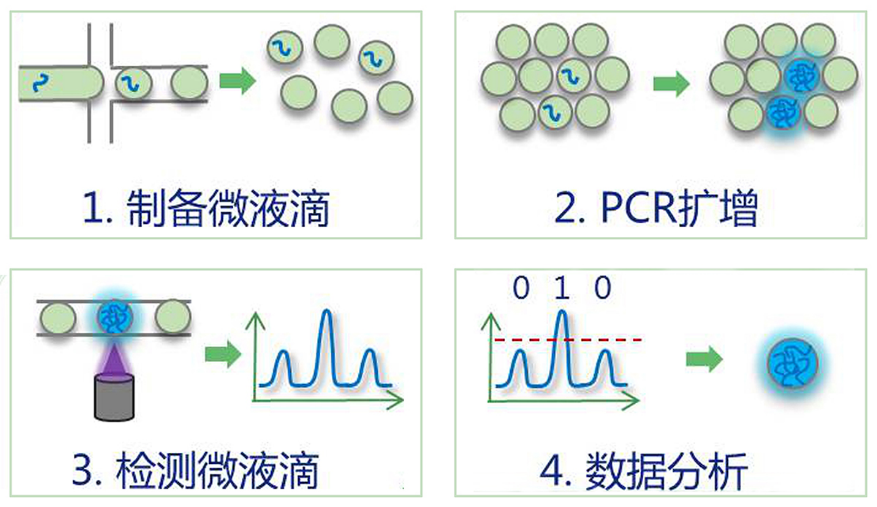
图12. 数字PCR流程
产品推荐
|
产品定位 |
产品名称 |
货号 |
应用场景 |
|
高保真PCR |
10148ES |
基因克隆、平末端克隆、定点突变 |
|
|
10149ES |
|||
|
10153ES |
分子克隆、测序、定点突变等高保真性PCR |
||
|
10154ES |
|||
|
常规PCR |
10101ES |
TA克隆、菌落PCR、基因型鉴定 |
|
|
10102ES |
|||
|
快速PCR |
10157ES |
基因快速扩增 |
|
|
长片段PCR |
10158ES |
适用于5kb以上的长片段,最长可扩增40kb |
|
|
10159ES |
|||
|
PAGE PCR mix |
10160ES |
小片段DNA, PAGE电泳 |
|
|
直扩PCR |
10184ES |
动物组织直接PCR |
|
|
10187ES |
植物组织直接PCR |
||
|
10188ES |
血液直接PCR |
||
|
10189ES |
小鼠组织直接PCR |
点击产品名称查看详情

